Deep Dive: Advanced Neuron Querying#
This tutorial explores the powerful and flexible querying capabilities of CRANTpy. We’ll go beyond simple filters and dive into:
Complex, multi-parameter queries
Using regular expressions for flexible matching
Inspecting available data to build your queries
Analyzing populations of neurons returned from queries
Let’s get started by setting up our environment.
# Import CRANTpy and other necessary libraries
import crantpy as cp
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
# Set up logging to see progress
cp.set_logging_level("WARNING") # Quieter for this tutorial
print("CRANTpy loaded successfully!")
print(f"Default dataset: {cp.CRANT_DEFAULT_DATASET}")
CRANTpy loaded successfully!
Default dataset: latest
1. Authentication Setup#
First, ensure you are authenticated with the CAVE service.
# Generate and save authentication token (uncomment if first time)
# cp.generate_cave_token(save=True)
# Test connection
try:
client = cp.get_cave_client()
print(f"Successfully connected to datastack: {client.datastack_name}")
print(f"Server: {client.server_address}")
except Exception as e:
print(f"Connection failed: {e}")
print("Please run: cp.generate_cave_token(save=True)")
Successfully connected to datastack: kronauer_ant
Server: https://proofreading.zetta.ai
2. Exploring Available Annotation Data#
To build powerful queries, you first need to know what data you can filter on. Let’s inspect the available annotation fields and their values.
# Get all available annotation fields
available_fields = cp.NeuronCriteria.available_fields()
print(f"Available annotation fields ({len(available_fields)}):")
for field in sorted(available_fields):
print(f" - {field}")
Available annotation fields (25):
- alternative_names
- annotator_notes
- cave_table
- cell_class
- cell_instance
- cell_subtype
- cell_type
- date_proofread
- flow
- hemilineage
- known_nt
- known_nt_source
- nerve
- ngl_link
- nucleus_id
- proofread
- proofreader_notes
- region
- root_id
- side
- status
- super_class
- tract
- user_annotator
- user_proofreader
# Get an overview of the entire dataset annotations
all_annotations = cp.get_all_seatable_annotations()
print(f"Total neurons in dataset: {len(all_annotations):,}")
print(f"Dataset shape: {all_annotations.shape}")
print("\nFirst few rows:")
display(all_annotations.head())
Total neurons in dataset: 6,166
Dataset shape: (6166, 30)
First few rows:
| root_id | root_id_processed | supervoxel_id | position | nucleus_id | nucleus_position | root_position | cave_table | proofread | status | ... | cell_subtype | cell_instance | known_nt | known_nt_source | alternative_names | annotator_notes | user_annotator | user_proofreader | ngl_link | date_proofread | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 576460752700282748 | None | 74170512125421134 | [32782, 30214, 1532] | 72691394107456688 | [37306, 31317, 1405] | [37306, 31317, 1405] | None | False | [DAMAGED, PARTIALLY_PROOFREAD, TRACING_ISSUE] | ... | None | None | acetylcholine | Tanaka et al., 2012 (immuno, mALT, drosophila ... | None | None | [lindsey_lopes] | [lindsey_lopes] | https://spelunker.cave-explorer.org/#!middleau... | None |
| 1 | 576460752681552812 | None | 74100212167609429 | [32121, 31509, 1702] | 72621025497478503 | [36772, 28974, 1953] | [36772, 28974, 1953] | None | True | [BACKBONE_PROOFREAD] | ... | None | None | acetylcholine | Tanaka et al., 2012 (immuno, mALT, drosophila ... | None | None | [lindsey_lopes] | [lindsey_lopes] | https://spelunker.cave-explorer.org/#!middleau... | None |
| 2 | 576460752666303418 | None | 74169069687405059 | [33220, 8787, 4046] | 72620682436952978 | [33727, 8389, 4054] | [33727, 8389, 4054] | None | False | [PARTIALLY_PROOFREAD, TRACING_ISSUE] | ... | None | None | acetylcholine, sNPF | Barnstedt et al. 2016 (immuno, KCs, drosophila... | None | None | [lindsey_lopes] | [lindsey_lopes] | https://spelunker.cave-explorer.org/#!middleau... | None |
| 3 | 576460752722405178 | None | 74100212167388307 | [32442, 31693, 1618] | 72621025497491534 | [36266, 31490, 2021] | [36266, 31490, 2021] | None | False | [PARTIALLY_PROOFREAD, TRACING_ISSUE] | ... | None | None | acetylcholine | Tanaka et al., 2012 (immuno, mALT, drosophila ... | None | None | [lindsey_lopes] | [lindsey_lopes] | https://spelunker.cave-explorer.org/#!middleau... | None |
| 4 | 576460752773799604 | None | 74100280887219649 | [32484, 32119, 1756] | 72691394308791443 | [37240, 29878, 2178] | [37240, 29878, 2178] | None | True | [BACKBONE_PROOFREAD] | ... | None | None | acetylcholine | Tanaka et al., 2012 (immuno, mALT, drosophila ... | None | None | [lindsey_lopes] | [lindsey_lopes] | https://spelunker.cave-explorer.org/#!middleau... | None |
5 rows × 30 columns
Exploring Data Distribution#
Let’s visualize some of the key annotation fields to understand the dataset better.
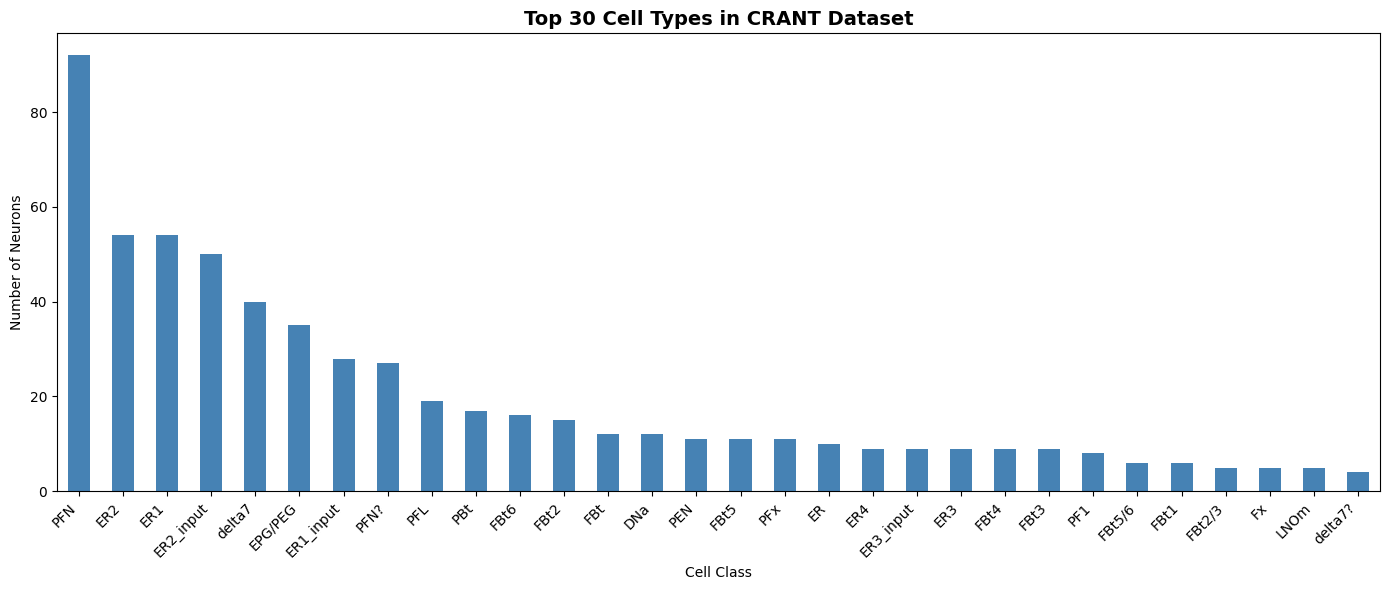
# Explore the distribution of cell types
cell_type_counts = all_annotations['cell_type'].value_counts()
print("Top 30 annotated cell types:")
print(cell_type_counts.head(30))
# Visualize
plt.figure(figsize=(14, 6))
cell_type_counts.head(30).plot(kind='bar', color='steelblue')
plt.title('Top 30 Cell Types in CRANT Dataset', fontsize=14, fontweight='bold')
plt.xlabel('Cell Class')
plt.ylabel('Number of Neurons')
plt.xticks(rotation=45, ha='right')
plt.tight_layout()
plt.show()
Top 30 annotated cell types:
cell_type
PFN 92
ER2 54
ER1 54
ER2_input 50
delta7 40
EPG/PEG 35
ER1_input 28
PFN? 27
PFL 19
PBt 17
FBt6 16
FBt2 15
FBt 12
DNa 12
PEN 11
FBt5 11
PFx 11
ER 10
ER4 9
ER3_input 9
ER3 9
FBt4 9
FBt3 9
PF1 8
FBt5/6 6
FBt1 6
FBt2/3 5
Fx 5
LNOm 5
delta7? 4
Name: count, dtype: int64
3. Basic Querying with NeuronCriteria#
The NeuronCriteria class is the main interface for filtering neurons. Let’s start with simple queries.
Simple Single-Criterion Queries#
The most basic query is filtering by a single field.
# Query neurons by cell class
olfactory_projection_neurons = cp.NeuronCriteria(cell_class='olfactory_projection_neuron')
opn_ids = olfactory_projection_neurons.get_roots()
print(f"Found {len(opn_ids)} olfactory projection neurons")
# Query neurons by side
left_neurons = cp.NeuronCriteria(side='left')
left_ids = left_neurons.get_roots()
print(f"Found {len(left_ids)} left-side neurons")
# Query neurons by tract
malt_neurons = cp.NeuronCriteria(tract='mALT')
malt_ids = malt_neurons.get_roots()
print(f"Found {len(malt_ids)} neurons in mALT tract")
Found 107 olfactory projection neurons
Found 3648 left-side neurons
Found 2275 neurons in mALT tract
Multi-Criterion Queries (Logical AND)#
You can combine multiple criteria - the query will return neurons matching all criteria (logical AND).
# Combine cell class and side
left_opn = cp.NeuronCriteria(
cell_class='olfactory_projection_neuron',
side='left'
)
left_opn_ids = left_opn.get_roots()
print(f"Found {len(left_opn_ids)} left olfactory projection neurons")
# Combine region, side, and proofread status
mb_left_proofread = cp.NeuronCriteria(
region='MB',
side='left',
proofread=True
)
mb_left_proofread_ids = mb_left_proofread.get_roots()
print(f"Found {len(mb_left_proofread_ids)} proofread left mushroom body neurons")
Found 37 left olfactory projection neurons
Found 30 proofread left mushroom body neurons
4. Advanced Filtering Options#
CRANTpy offers powerful filtering options for complex queries.
Using Regular Expressions#
Regular expressions (regex=True) enable powerful pattern matching for finding related groups of neurons.
# Find all ExR subtypes using regex
ExR_subtypes = cp.NeuronCriteria(
cell_type='ExR', # Matches ExR_a, ExR_b, etc.
regex=True
)
ExR_subtype_ids = ExR_subtypes.get_roots()
print(f"Found {len(ExR_subtype_ids)} ExR subtype neurons")
# Get their annotations to see the specific types
exR_annotations = cp.get_annotations(ExR_subtypes)
print("\nExR subtypes found:")
print(exR_annotations['cell_type'].value_counts())
# More complex regex: Find all tangential neurons (FBt or PBt)
all_tangential = cp.NeuronCriteria(
cell_type='[FBt|PBt]', # Matches FBt or PBt
regex=True
)
all_tangential_ids = all_tangential.get_roots()
print(f"\nFound {len(all_tangential_ids)} tangential neurons (FBt/PBt)")
Found 6 ExR subtype neurons
2025-10-10 07:58:43 - WARNING - Failed to update 2 annotation root ID(s). These IDs may no longer exist.
ExR subtypes found:
cell_type
ExR1 4
ExR2 2
Name: count, dtype: int64
Found 450 tangential neurons (FBt/PBt)
Filtering with Lists: match_all Parameter#
When filtering by a list of values on fields that contain lists (like status or region), the match_all parameter controls the matching behavior.
# match_all=False (default): neurons with ANY of the specified statuses
any_status = cp.NeuronCriteria(
status=['BACKBONE_PROOFREAD', 'PRELIM_PROOFREAD'],
match_all=False # This is the default
)
any_status_ids = any_status.get_roots()
print(f"Neurons with ANY of the specified statuses: {len(any_status_ids)}")
# match_all=True: neurons with ALL of the specified statuses
all_statuses = cp.NeuronCriteria(
status=['DAMAGED', 'TRACING_ISSUE'],
match_all=True
)
try:
all_statuses_ids = all_statuses.get_roots()
print(f"Neurons with ALL of the specified statuses: {len(all_statuses_ids)}")
except Exception as e:
print(f"Query returned: {e}")
# Practical example: neurons in multiple regions
multi_region = cp.NeuronCriteria(
region=['MB', 'AL'], # Neurons that innervate both regions
match_all=True
)
try:
multi_region_ids = multi_region.get_roots()
print(f"\nNeurons in BOTH MB and AL: {len(multi_region_ids)}")
except Exception as e:
print(f"\nQuery returned: {e}")
Neurons with ANY of the specified statuses: 715
Neurons with ALL of the specified statuses: 2
Neurons in BOTH MB and AL: 128
Substring Matching: exact Parameter#
By default, queries match values exactly. Setting exact=False enables substring matching.
# exact=True (default): exact matching only
exact_match = cp.NeuronCriteria(
cell_class='olfactory_projection_neuron',
exact=True
)
exact_ids = exact_match.get_roots()
print(f"Exact match for 'olfactory_projection_neuron': {len(exact_ids)}")
# exact=False: substring matching
substring_match = cp.NeuronCriteria(
cell_class='projection', # Will match any cell_class containing 'projection'
exact=False
)
substring_ids = substring_match.get_roots()
print(f"Substring match for 'projection': {len(substring_ids)}")
# See what was matched
substring_annotations = cp.get_annotations(substring_match)
print("\nCell classes matched (substring 'projection'):")
print(substring_annotations['cell_class'].value_counts())
Exact match for 'olfactory_projection_neuron': 107
Substring match for 'projection': 107
Cell classes matched (substring 'projection'):
cell_class
olfactory_projection_neuron 109
spiny_kenyon_cell 1
Name: count, dtype: int64
Cell classes matched (substring 'projection'):
cell_class
olfactory_projection_neuron 109
spiny_kenyon_cell 1
Name: count, dtype: int64
Case-Sensitive Matching#
By default, string matching is case-insensitive. Use case=True for case-sensitive matching (only applies when regex=True).
# Case-insensitive (default)
case_insensitive = cp.NeuronCriteria(
cell_type='exr', # Will match ExR, exr, ExR_a, etc.
regex=True,
case=False # This is the default
)
case_insensitive_ids = case_insensitive.get_roots()
print(f"Case-insensitive match for 'exr': {len(case_insensitive_ids)}")
# Case-sensitive
case_sensitive = cp.NeuronCriteria(
cell_type='exr', # Will only match exact case
regex=True,
case=True
)
try:
case_sensitive_ids = case_sensitive.get_roots()
print(f"Case-sensitive match for 'exr': {len(case_sensitive_ids)}")
except Exception as e:
print(f"Case-sensitive match returned: {e}")
Case-insensitive match for 'exr': 6
Case-sensitive match returned: No neurons found matching the given criteria.
5. Working with Annotations#
Beyond just getting root IDs, you can fetch full annotation data for neurons.
# Get annotations for a NeuronCriteria object
mb_neurons = cp.NeuronCriteria(region='MB')
mb_annotations = cp.get_annotations(mb_neurons)
print(f"Retrieved annotations for {len(mb_annotations)} MB neurons")
print(f"\nColumns available: {list(mb_annotations.columns)}")
print("\nFirst few rows:")
display(mb_annotations[['root_id', 'cell_class', 'cell_type', 'side', 'region']].head())
# Get annotations for specific root IDs
sample_ids = opn_ids[:5]
sample_annotations = cp.get_annotations(sample_ids)
print(f"\nAnnotations for {len(sample_annotations)} specific neurons:")
display(sample_annotations[['root_id', 'cell_class', 'cell_type', 'tract']])
Retrieved annotations for 323 MB neurons
Columns available: ['root_id', 'root_id_processed', 'supervoxel_id', 'position', 'nucleus_id', 'nucleus_position', 'root_position', 'cave_table', 'proofread', 'status', 'region', 'proofreader_notes', 'side', 'nerve', 'tract', 'hemilineage', 'flow', 'super_class', 'cell_class', 'cell_type', 'cell_subtype', 'cell_instance', 'known_nt', 'known_nt_source', 'alternative_names', 'annotator_notes', 'user_annotator', 'user_proofreader', 'ngl_link', 'date_proofread']
First few rows:
| root_id | cell_class | cell_type | side | region | |
|---|---|---|---|---|---|
| 0 | 576460752700282748 | olfactory_projection_neuron | None | left | [LH, AL, MB] |
| 1 | 576460752681552812 | olfactory_projection_neuron | None | left | [LH, AL, MB] |
| 2 | 576460752666303418 | spiny_kenyon_cell | None | right | [MB] |
| 3 | 576460752722405178 | olfactory_projection_neuron | None | left | [LH, AL, MB] |
| 4 | 576460752773799604 | olfactory_projection_neuron | None | right | [LH, AL, MB] |
Annotations for 10 specific neurons:
| root_id | cell_class | cell_type | tract | |
|---|---|---|---|---|
| 0 | 576460752700282748 | olfactory_projection_neuron | None | mALT |
| 1 | 576460752681552812 | olfactory_projection_neuron | None | mALT |
| 2 | 576460752722405178 | olfactory_projection_neuron | None | mALT |
| 3 | 576460752773799604 | olfactory_projection_neuron | None | mALT |
| 4 | 576460752656800770 | olfactory_projection_neuron | None | lALT |
| 5 | 576460752722405178 | spiny_kenyon_cell | None | None |
| 6 | 576460752681552812 | None | None | mALT |
| 7 | 576460752722405178 | None | None | mALT |
| 8 | 576460752773799604 | None | None | mALT |
| 9 | 576460752700282748 | None | None | mALT |
Automatic ID Updating#
CRANTpy automatically handles segmentation edits through the update_ids parameter. When neurons are edited in the segmentation, their root IDs change. This can cause mismatches between your query IDs and annotation IDs.
# By default, both NeuronCriteria and get_annotations update IDs automatically
# Example 1: NeuronCriteria automatically updates returned IDs
nc = cp.NeuronCriteria(cell_class='olfactory_projection_neuron')
current_ids = nc.get_roots() # These are the latest root IDs
print(f"NeuronCriteria returned {len(current_ids)} neurons with updated IDs")
# Example 2: get_annotations updates annotation IDs before matching
# This ensures that even if you have a newer root ID, it will match
# against the updated annotation IDs
sample_id = int(current_ids[0]) # Convert numpy int64 to Python int
annotations = cp.get_annotations(sample_id) # update_ids=True by default
print(f"\nSuccessfully retrieved annotations for ID {sample_id}")
print(f"Cell class: {annotations.iloc[0]['cell_class']}")
# You can disable automatic updating if needed
nc_no_update = cp.NeuronCriteria(cell_class='olfactory_projection_neuron', update_ids=False)
old_style_ids = nc_no_update.get_roots() # IDs as stored in annotations
print(f"\nWithout update_ids: {len(old_style_ids)} neurons")
# Similarly for get_annotations
annotations_no_update = cp.get_annotations(sample_id, update_ids=False)
print(f"Annotations retrieved without ID updating")
NeuronCriteria returned 107 neurons with updated IDs
Successfully retrieved annotations for ID 576460752700282748
Cell class: olfactory_projection_neuron
Without update_ids: 107 neurons
Annotations retrieved without ID updating
Why is this important?
When the segmentation is edited (e.g., neurons are merged or split), root IDs change. Without automatic updating:
Your queries might use outdated IDs that no longer exist
Annotations might contain old IDs that don’t match your current data
You’d need to manually track and update IDs
With automatic updating enabled (the default):
NeuronCriteria.get_roots()returns the latest root IDsget_annotations()updates annotation IDs before matchingEverything stays synchronized automatically
Per-ID caching makes this efficient
6. Checking Proofread Status#
You can quickly check which neurons are proofread using the is_proofread function.
# Check proofread status for a list of neurons
sample_ids = opn_ids[:10]
proofread_status = cp.is_proofread(sample_ids)
print(f"Checking proofread status for {len(sample_ids)} neurons:")
for neuron_id, is_pr in zip(sample_ids, proofread_status):
status = "✓ Proofread" if is_pr else "✗ Not proofread"
print(f" {neuron_id}: {status}")
print(f"\nSummary: {np.sum(proofread_status)}/{len(proofread_status)} are proofread")
# You can also check using NeuronCriteria
proofread_status_criteria = cp.is_proofread(olfactory_projection_neurons)
print(f"\nOf all {len(olfactory_projection_neurons)} OPNs, {np.sum(proofread_status_criteria)} are proofread")
Checking proofread status for 10 neurons:
576460752700282748: ✗ Not proofread
576460752681552812: ✓ Proofread
576460752722405178: ✗ Not proofread
576460752773799604: ✓ Proofread
576460752656800770: ✓ Proofread
576460752722298426: ✗ Not proofread
576460752768215072: ✗ Not proofread
576460752680204173: ✓ Proofread
576460752728003009: ✓ Proofread
576460752780951210: ✗ Not proofread
Summary: 5/10 are proofread
Of all 107 OPNs, 65 are proofread
7. NeuronCriteria as Iterable and Container#
NeuronCriteria objects support Python’s iterator and container protocols, making them very convenient to use.
# Get length
print(f"Number of OPNs: {len(olfactory_projection_neurons)}")
# Iterate over root IDs
print("\nFirst 5 OPN root IDs (using iteration):")
for i, root_id in enumerate(olfactory_projection_neurons):
if i >= 5:
break
print(f" {root_id}")
# Check membership
sample_id = opn_ids[0]
is_member = sample_id in olfactory_projection_neurons
print(f"\nIs {sample_id} an OPN? {is_member}")
# Check a neuron that's not an OPN
non_opn_id = left_ids[0] # A random left neuron
is_member = non_opn_id in olfactory_projection_neurons
print(f"Is {non_opn_id} an OPN? {is_member}")
Number of OPNs: 107
First 5 OPN root IDs (using iteration):
576460752700282748
576460752681552812
576460752722405178
576460752773799604
576460752656800770
Is 576460752700282748 an OPN? True
Is 576460752700282748 an OPN? True
8. Performance and Caching#
CRANTpy caches annotation data to improve performance. You can control this behavior.
import time
# First query - may take longer (fetching from Seatable)
start = time.time()
query1 = cp.NeuronCriteria(cell_class='olfactory_projection_neuron', clear_cache=True)
ids1 = query1.get_roots()
time1 = time.time() - start
print(f"First query (no cache): {time1:.3f} seconds")
# Second query - should be faster (using cached data)
start = time.time()
query2 = cp.NeuronCriteria(cell_class='olfactory_projection_neuron')
ids2 = query2.get_roots()
time2 = time.time() - start
print(f"Second query (cached): {time2:.3f} seconds")
# Force refresh from Seatable
start = time.time()
query3 = cp.NeuronCriteria(cell_class='olfactory_projection_neuron', clear_cache=True)
ids3 = query3.get_roots()
time3 = time.time() - start
print(f"Query with cache refresh: {time3:.3f} seconds")
First query (no cache): 2.984 seconds
Second query (cached): 0.006 seconds
Query with cache refresh: 1.749 seconds
Query with cache refresh: 1.749 seconds
9. Analyzing Query Results#
Once you have a set of neurons, you can perform various analyses on their annotations.
# Analyze connectivity patterns by tract
tracts = all_annotations['tract'].dropna().unique()
print(f"Found {len(tracts)} tracts: {list(tracts)[:10]}...") # Show first 10
# Analyze each tract
tract_analysis = []
for tract in tracts[:8]: # Analyze first 8 tracts for demonstration
tract_neurons = cp.NeuronCriteria(tract=tract)
tract_ids = tract_neurons.get_roots()
tract_annotations = cp.get_annotations(tract_neurons)
# Count unique cell classes
n_cell_classes = len(tract_annotations['cell_class'].unique())
n_cell_types = len(tract_annotations['cell_type'].dropna().unique())
# Count proofread neurons
n_proofread = tract_annotations['proofread'].sum() if 'proofread' in tract_annotations.columns else 0
tract_analysis.append({
'tract': tract,
'n_neurons': len(tract_ids),
'n_cell_classes': n_cell_classes,
'n_cell_types': n_cell_types,
'n_proofread': n_proofread,
'pct_proofread': f"{100 * n_proofread / len(tract_ids):.1f}%"
})
tract_df = pd.DataFrame(tract_analysis)
print("\nTract analysis:")
display(tract_df)
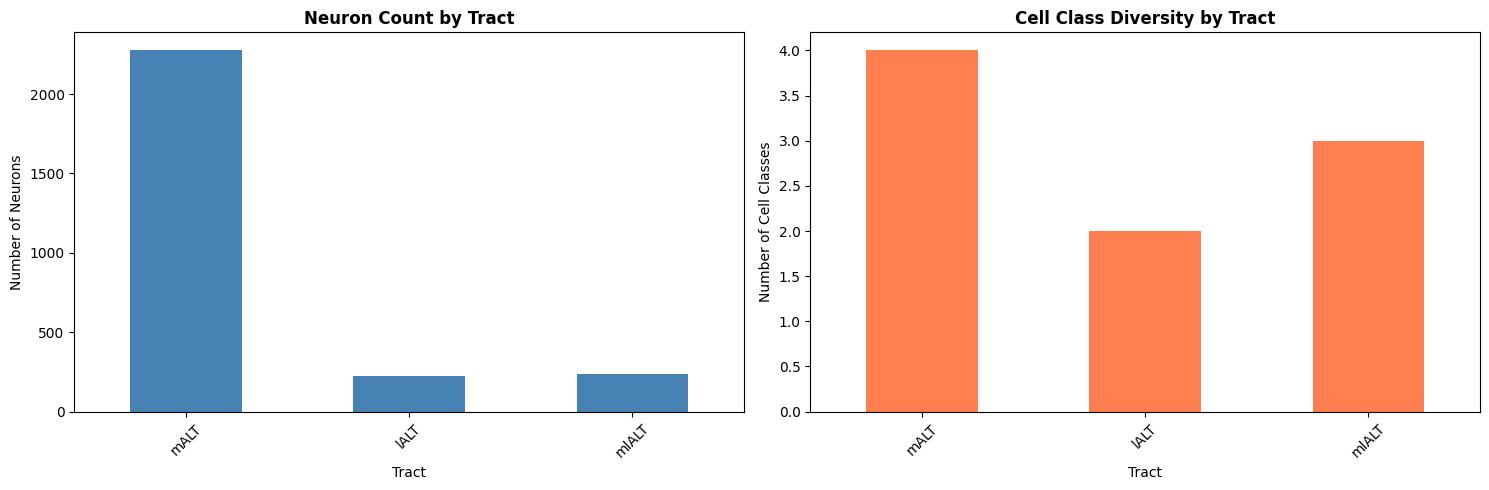
# Visualize tract composition
fig, (ax1, ax2) = plt.subplots(1, 2, figsize=(15, 5))
tract_df.plot(x='tract', y='n_neurons', kind='bar', ax=ax1, color='steelblue', legend=False)
ax1.set_title('Neuron Count by Tract', fontsize=12, fontweight='bold')
ax1.set_xlabel('Tract')
ax1.set_ylabel('Number of Neurons')
ax1.tick_params(axis='x', rotation=45)
tract_df.plot(x='tract', y='n_cell_classes', kind='bar', ax=ax2, color='coral', legend=False)
ax2.set_title('Cell Class Diversity by Tract', fontsize=12, fontweight='bold')
ax2.set_xlabel('Tract')
ax2.set_ylabel('Number of Cell Classes')
ax2.tick_params(axis='x', rotation=45)
plt.tight_layout()
plt.show()
Found 3 tracts: ['mALT', 'lALT', 'mlALT']...
2025-10-10 08:02:04 - WARNING - Failed to update 2 annotation root ID(s). These IDs may no longer exist.
Tract analysis:
| tract | n_neurons | n_cell_classes | n_cell_types | n_proofread | pct_proofread | |
|---|---|---|---|---|---|---|
| 0 | mALT | 2275 | 4 | 0 | 41 | 1.8% |
| 1 | lALT | 227 | 2 | 0 | 29 | 12.8% |
| 2 | mlALT | 235 | 3 | 0 | 3 | 1.3% |
Cross-Analysis: Comparing Populations#
# Compare left vs right neurons
left_neurons = cp.NeuronCriteria(side='left')
right_neurons = cp.NeuronCriteria(side='right')
left_ann = cp.get_annotations(left_neurons)
right_ann = cp.get_annotations(right_neurons)
print(f"Left side: {len(left_ann)} neurons")
print(f"Right side: {len(right_ann)} neurons")
# Compare cell class distribution
print("\nTop 5 cell classes on left:")
print(left_ann['cell_class'].value_counts().head())
print("\nTop 5 cell classes on right:")
print(right_ann['cell_class'].value_counts().head())
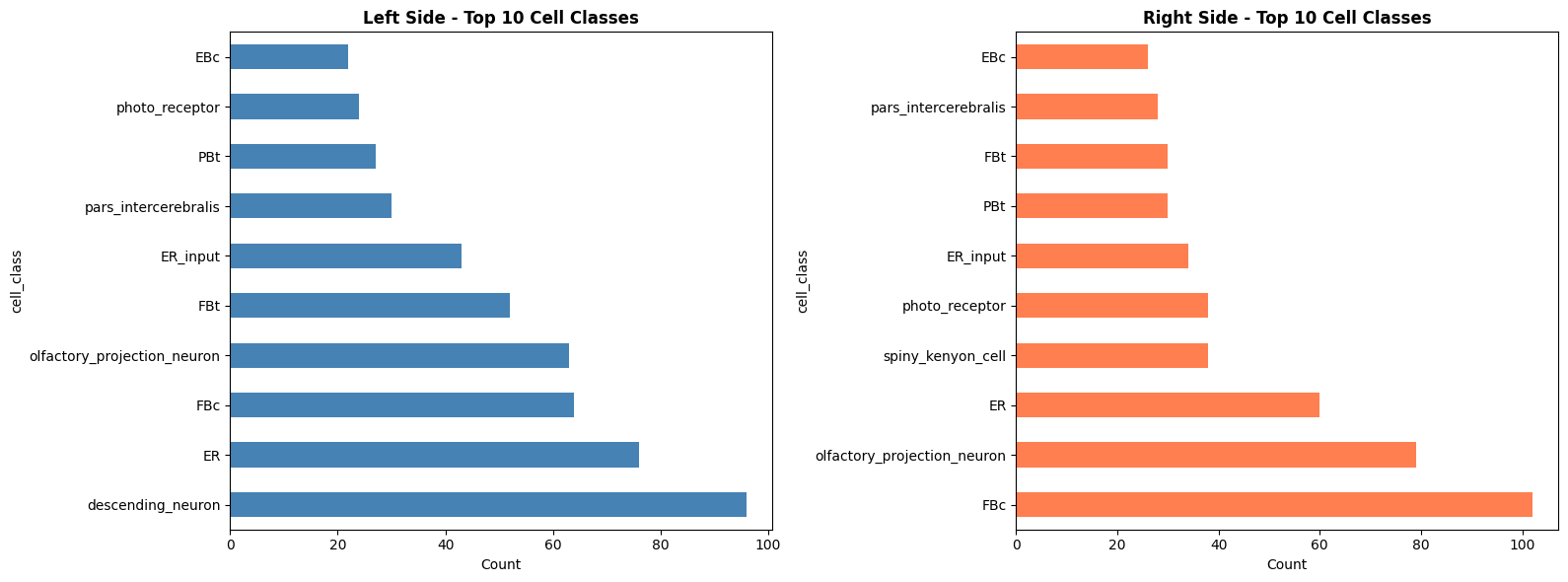
# Visualize comparison
fig, axes = plt.subplots(1, 2, figsize=(16, 6))
left_ann['cell_class'].value_counts().head(10).plot(kind='barh', ax=axes[0], color='steelblue')
axes[0].set_title('Left Side - Top 10 Cell Classes', fontsize=12, fontweight='bold')
axes[0].set_xlabel('Count')
right_ann['cell_class'].value_counts().head(10).plot(kind='barh', ax=axes[1], color='coral')
axes[1].set_title('Right Side - Top 10 Cell Classes', fontsize=12, fontweight='bold')
axes[1].set_xlabel('Count')
plt.tight_layout()
plt.show()
Left side: 3792 neurons
Right side: 1897 neurons
Top 5 cell classes on left:
cell_class
descending_neuron 96
ER 76
FBc 64
olfactory_projection_neuron 63
FBt 52
Name: count, dtype: int64
Top 5 cell classes on right:
cell_class
FBc 102
olfactory_projection_neuron 79
ER 60
spiny_kenyon_cell 38
photo_receptor 38
Name: count, dtype: int64
10. Best Practices and Tips#
Here are some best practices for effective querying with CRANTpy.
1. Start Broad, Then Refine#
Begin with broad queries and progressively add criteria to narrow down your results.
# Step 1: Broad query
step1 = cp.NeuronCriteria(region='CX')
print(f"Step 1 - All CX neurons: {len(step1)}")
# Step 2: Add side constraint
step2 = cp.NeuronCriteria(region='CX', side='left')
print(f"Step 2 - Left CX neurons: {len(step2)}")
# Step 3: Add cell class
step3 = cp.NeuronCriteria(region='CX', side='left', cell_class='ER')
print(f"Step 3 - Left CX ER neurons: {len(step3)}")
# Step 4: Add proofread constraint
step4 = cp.NeuronCriteria(region='CX', side='left', cell_class='ER', proofread=True)
print(f"Step 4 - Proofread left CX ER neurons: {len(step4)}")
Step 1 - All CX neurons: 656
Step 2 - Left CX neurons: 298
Step 3 - Left CX ER neurons: 70
Step 4 - Proofread left CX ER neurons: 59
2. Use verbose=True for Debugging#
Enable verbose mode to see what’s happening during queries.
cp.set_logging_level("DEBUG") # this needs to be updated to see messages
# Without verbose (default)
query_quiet = cp.NeuronCriteria(cell_class='olfactory_projection_neuron', side='left')
ids_quiet = query_quiet.get_roots()
# With verbose
query_verbose = cp.NeuronCriteria(
cell_class='olfactory_projection_neuron',
side='left',
verbose=True
)
ids_verbose = query_verbose.get_roots()
cp.set_logging_level("WARNING")
2025-10-10 08:02:05 - DEBUG - Generated cache key: latest for seatable_annotations
2025-10-10 08:02:05 - DEBUG - Cache hit for seatable_annotations with key: latest
2025-10-10 08:02:05 - DEBUG - Using cached seatable_annotations.
2025-10-10 08:02:05 - DEBUG - update_ids_cache: 37 cached, 0 uncached
2025-10-10 08:02:05 - DEBUG - Using fully cached results from update_ids_cache
2025-10-10 08:02:05 - DEBUG - Cache hit for seatable_annotations with key: latest
2025-10-10 08:02:05 - DEBUG - Using cached seatable_annotations.
2025-10-10 08:02:05 - DEBUG - update_ids_cache: 37 cached, 0 uncached
2025-10-10 08:02:05 - DEBUG - Using fully cached results from update_ids_cache
2025-10-10 08:02:05 - DEBUG - Generated cache key: latest for seatable_annotations
2025-10-10 08:02:05 - DEBUG - Cache hit for seatable_annotations with key: latest
2025-10-10 08:02:05 - DEBUG - Using cached seatable_annotations.
2025-10-10 08:02:05 - INFO - Found 37 neurons matching the given criteria.
2025-10-10 08:02:05 - DEBUG - update_ids_cache: 37 cached, 0 uncached
2025-10-10 08:02:05 - DEBUG - Using fully cached results from update_ids_cache
2025-10-10 08:02:05 - DEBUG - Generated cache key: latest for seatable_annotations
2025-10-10 08:02:05 - DEBUG - Cache hit for seatable_annotations with key: latest
2025-10-10 08:02:05 - DEBUG - Using cached seatable_annotations.
2025-10-10 08:02:05 - INFO - Found 37 neurons matching the given criteria.
2025-10-10 08:02:05 - DEBUG - update_ids_cache: 37 cached, 0 uncached
2025-10-10 08:02:05 - DEBUG - Using fully cached results from update_ids_cache
3. Inspect Available Values Before Querying#
Check what values exist in your fields of interest before building queries.
# Check what tracts are available
print("Available tracts:")
print(all_annotations['tract'].dropna().unique())
# Check what statuses exist
print("\nAvailable statuses (flatten lists):")
status_lists = all_annotations['status'].dropna().tolist()
flat_statuses = set([status for sublist in status_lists for status in (sublist if isinstance(sublist, list) else [sublist])])
print(flat_statuses)
# Check cell types for a specific cell class
fbt_neurons_check = cp.NeuronCriteria(cell_class='FBt')
fbt_ann_check = cp.get_annotations(fbt_neurons_check)
print("\nCell types for mushroom_body_intrinsic_neuron:")
print(fbt_ann_check['cell_type'].value_counts())
Available tracts:
['mALT' 'lALT' 'mlALT']
Available statuses (flatten lists):
{'MISSING_SOMA', 'empty', 'NA', 'MERGE_ERROR', 'MERGE_MONSTER', 'DUPLICATED', 'TO_PROOFREAD', 'BACKBONE_PROOFREAD', 'PARTIALLY_PROOFREAD', 'DAMAGED', 'TRACING_ISSUE', 'THOROUGHLY_PROOFREAD'}
Cell types for mushroom_body_intrinsic_neuron:
cell_type
FBt6 16
FBt2 15
FBt 12
FBt5 11
FBt4 9
FBt3 9
FBt1 6
FBt5/6 6
FBt2/3 5
FBt1/2 2
Name: count, dtype: int64
Summary#
In this comprehensive deep dive, you learned:
✅ NeuronCriteria fundamentals - Basic and multi-criterion queries
✅ Advanced filtering options - regex, match_all, exact, case parameters
✅ Working with annotations - Fetching and analyzing neuron metadata
✅ Proofread status checking - Identifying which neurons are proofread
✅ Iterator/container protocols - Using NeuronCriteria as iterable objects
✅ Performance optimization - Understanding and controlling caching behavior
✅ Population analysis - Comparing and analyzing query results
✅ Best practices - Strategies for efficient and effective querying
Key Takeaways#
Combine criteria: Use multiple parameters to precisely target neuron populations
Leverage regex: Pattern matching is powerful for finding related neuron groups
Understand match_all: Control how lists are matched (ANY vs ALL)
Inspect first: Always explore available data before building complex queries
Use annotations: Don’t just get IDs - analyze the full metadata